PT試薬の標準化 PT Standarization
A 標準化の意義と目的
(1) はじめに
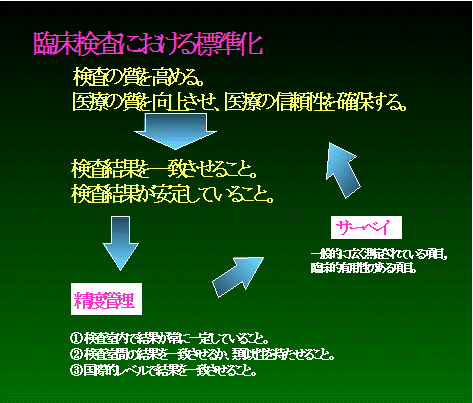
結果が安定であるためには、まず、1つの検査室において継続して安定した結果でなければなりませんし、また、各検査室間で結果が一致することが必要です。
これらの作業をサポートする手段として、全国的、あるいは全世界的にサーベイが実施されています。現在、我々は各種のサーベイに参加し、自分の検査結果が、あるいは自検査室が全体レベルのどの位置にあるのかを知ることができます。それによって、改善の機会が与えられていることになります。
しかしながら、サーベイにおいては臨床検査で実施される項目を全部実行することは不可能です。よって、サーベイでは一般的に広く測定されている項目(ポピュラーな項目)であって、しかも臨床的意義の高いものを選択して実施することになります。サーベイでは、一般的、且つ、有効な項目から統一し、やがては臨床検査全体の統一を目指しています。
凝固分野では、最もポプラーな測定項目であるPTをターゲットとして、現在、PTを標準化することにより、データの統一を図り、やがては凝固検査全体の統一化を図ろうとしています。凝固分野での標準化活動はスタートラインに着き、“やっと一歩踏み出した”と言うところでしょう。
A-2 PTの表現方法
PTの表現方法の表現方法としては、以下のようなものが挙げられます。
① 秒数
② 正常/異常
③ 異常/正常
④ 活性%の表示
⑤ INR表示
PT結果を報告する方法として、最初に利用された表現方法は ① 秒数で、これは現在でも使用して いる施設があります。測定結果は概念的(感覚的)に異常の程度を推定することができます。また、継続して 観察すれば病態の経過を知ることができます。しかし、測定結果は試薬ロットによって変動し、精密に比較す るには問題があります。
②および、③ は秒数として概念的に把握するのではなく、より客観的な値として診るために工夫されました。
この方法では、正常/正常 = 1.0 に対して、比としての値が高く表示されるか(または低く表示されるか)
によって、正常と比較した異常の程度を知ることができます。この場合、試薬ロット間差の影響を受けにくく
なり、秒数に比べればより客観的な値となります。けれども、同一の検査室の場合には比較できますが、他施
設との比較、あるいは種類の異なる試薬を使用する場合では、比較できなくなります。
活性%を使用する方法は、今日、本邦では最も広く使用されています。使う試薬それぞれに検量線を作成す
るので、秒数や比が異なっても、正常は100%と算出され、異常はその程度に応じて低値に表示されます。この
結果、活性%表示は多様な試薬間の比較ができ、標準化の手段として優れた方法であると思われます。ただ、
検量線が湾曲すると言う欠点もあります。
INR表示は、世界的標準化の検討において考案された方法です。ワーファリン治療、あるいはそのモニタ リングにおいて、活性%表示では湾曲する部分を直線的に表示する方法として採用されています。
A-3 凝固分野における標準化の流れ
A-3-1 歴史的 経緯
さて、血液凝固分野における標準化の試みでは、プロトロンビン時間(PT)が標準化の対象になっています。 現在、PT測定は、1935年にA.J.Quickが1段法によるプロトロンビン時間測定方法を提唱したものを一般 的に使用していますが、標準化の議論が開始された1960年代に、これが世界的に普及していた最もポピュラーな 項目であったために標準化の項目となったと考えられます。逆に、標準化の試みはPT結果を統一するために始 まったとも言えます。
1960年代のPT測定は用手法で実施されていました。したがって、PTの測定結果は試薬あるいは検体(血 漿)の組み合わせで多様に変化しました。そこで、試薬を統一することで結果を一致させようとする方向と、検 体血漿を統一することで結果を一致させようとする方向の2つの試みが検討されました。
検体血漿を統一することによって標準化をおこなうという試みでは、アメリカを中心として広く議論され、 1971年イスタンブール(トルコ)でおこなわれた Maditerranean Congress on Thrombolism では「基本的に標 準血漿の概念は有用である」との認識が確認されました。この決議に基づいて標準血漿の作製作業が開始されま した。検討の過程では、「生血液や凍結血液では利用面で制約が多く凍結乾燥血漿を使用せざるを得ない」こと、 しかしながら、「凍結乾燥血漿では試薬との相性により結果が異なってくることがある」こと、「試薬の影響を 受けない血漿の開発が困難なこと」・・・・・・などの理由から標準血漿を特定し得ない状況になりました。その当時、 アメリカではOrtho社 と DADE社 の 2大メーカーがお互いに標準血漿を提供することを申し出ましたが、 むしろこれが設定作業の混乱に拍車をかけてしまったと考えられます。このため、現在でも国際的に合意された 標準血漿は存在しません。標準血漿として選定されているのは、フィブリノーゲン量や各凝固因子の活性量、 AT-Ⅲ・プラスミノーゲン・・・・・・などがNIBSC(イギリス)によって決定されています。が、PTやAP TTに関する標準血漿はありません。
試薬を統一することによって標準化をおこなうという試みでは、Dr.Poller(イギリス)によって精力的に進 められました。イギリスでは、「同じ製剤を使用して同じ検査結果を出す」試みが実際におこなわれ、 Dr.Pollerは輝かしい成果を残すことができました。今でもイギリスでは Manchester社のトロンボプラスチン 試薬が97%のシェアーを誇っているという報告もあります。しかしながら、この試みは同一試薬の消費量が多 すぎて、すぐに使い果たされてしまい、継続性の面で挫折を余儀なくされました。
そこで、次に、標準試薬の消費量を減らす工夫として、「一般試薬は標準試薬と比較した値を付けることに よって、標準試薬と同等の結果が得られるようにする」ことがを考えられました。これは、一般試薬を使用する 場合には、正常凝固時間に対して異常凝固時間の比(Ratio)をとり、BCT(British Comparative Thromboplastin) に対する傾きを修正する方法でした。
これにより、Biggs and Densonは、自ら作製したヒト脳注出のトロンボプラスチン製剤が、多くのPT試薬とグラフ上で補正できることを示しました。この概念の初期には、補正の方法は単純にRatioにIndexをかける方法だったために、正常~軽度異常の領域においては湾曲する現象が見られました。これは、臨床的に説明不能な矛盾でした。が、そのことが、逆に議論を活性化させ、ICSH(International Council for Standardization in Hematology)、及び、ICTH(International Committee on Thrombosis and Haemostasis)の設立につながりました。ICSHによるPT試験の標準法、ICTHによる各種の検討の結果、 Biggs and Denson の作製したロット67/40はWHOの国際標準トロンボプラスチンとして採用されました(1976年)。WHOは、テクニカルレポート(1983:ジュネーブ)を発表しています。次いで、ICSHとICTHの共同作業のもと、WHOはウシとウサギの脳由来の標準品が設定しています(1985年)。なお、この間、修正する方法は両対数グラフ上で補正する方法に代わり、補正値はISI(International Sensitivity Index)として使用されるようになっています。
設問(21): INRの計算方式はLog10を基本としている、か?
Yes or No
A-3-2 国際標準トロンボプラスチン試薬 の 設定
WHOが最初の国際標準トロンボプラスチンとして採用した試薬は、Biggs and Densonが作製したロット67/40です(1976)。この試薬は、これ以前の反省からすぐに門外不出の試薬としました。したがって、一般的に使用できる試薬を大量に準備する作業がはじまり、BCT/099が1978年に、1983年にはBCT/253がヒト脳由来のトロンボプラスチン試薬としてが設定されました。次いで1983年には、二次標準品として、ヒト、ウシ、ウサギ脳由来のトロンボプラスチン製剤が設定されました。これらは、一次標準品と直接比較検討されたISI値が付けられており、 ECの一部門であるBCRが認定標準材料としてその配布をおこなうことにより、世界的に比較検討できるシステムが完成しました。EC/BCRにおいて
BCT /009(ヒト脳、ISI = 1.048)は、CRM147として、
OBT / 79(ウシ脳、ISI = 1.011)は、CRM148として、
RBT / 79(ウサギ脳、ISI = 1.413)は、CRM149として登録・公開されています。
なお、これらの作業はICTHとICSHとの共同研究の成果であり、その報告は、Thrombosis and Haemostasis に掲載することで公開性を持たせています。これらの作業により、今日、我々はBCRから標準品を購入し、自施設での値が正しいか否かを判定することができるようになりました。
雑学13) : Biggs and Densonの内、Densonは複合因子TTO・HpT試薬のOEM供給元の社長です。
*国際標準品 の 経緯
1976 BCT67/40の設定(初代IRP、ISI = 1.00)
1983 BCT /253 (ヒト脳、ISI = 1.085)
1985 二次標品の設定
BCT /009 ヒト脳、ISI = 1.048
>
OBT / 79 ウシ脳、ISI = 1.011
RBT / 79 ウサギ脳、ISI = 1.413 ← CRM149
1991 CRM 149R ISI = 1.343
1995 CRM 149S ISI = 1.257
注)ICSH : International Council for Standardization in Hematology
(国際血液学標準化委員会)
ICTH : International Committee on Thrombosis and Haemostasis
(国際血栓止血学委員会)
BCT : British Comparative Thromboplastin(英国対照PT試薬)
英国血液学標準化委員会(British Committee for Standards in Haematology)
BCR : Reference Bureau of the European Union(欧州連合基準局)
IRP : International Reference Preparation(国際標準品)
CRM : Certified Reference Material
この後、BCRでは在庫の不足に伴い、さらなる標準試薬の設定と供給をせざるを得ない状況となって います。新しい標準試薬の設定には欧州の10ヶ所の検討施設で一次標準品および二次標準品を使っておこなわれ、 CRM149Rが1988年に、CRM149Sが1993年に設定されています(本邦において一般的に入手できるよう になったのはそれぞれ約3年後)。この結果、新たな標準品を設定するたびに、Golden Standardsは確実に減少し、 今や二次標準品でさえ試験に供することができない状態となっています。
雑学14) : ICTHからアナウンスされるものはだいたいThrombosis and Haemostasis(英文)に掲載されます。
A-4 WHOの勧告
3-A-4-1 テクニカルレポート の 要旨
以下の文献より
① WHO Expert Committee on Biological Stanndardization 33 report [WHO technical Report Series 687] 1983
② ICSH/ICTH Recommendations for Reporting Prothrombin Time In Oral Anticogulant Control T hrombos. Haemostas. 155-156 1985
③ Commission of the European Communities; BCR INFORMATION reference materials: The Certification of the second reference material for rabbit Thromboplastin CRM 149R 1988
④ Commission of the European Communities BCR INFORMATION reference materials:
The Certification of the third reference material for rabbit Thromboplastin CRM 149S 1994
⑤ Commission of the European Communities BCR INFORMATION reference materials: The Certification of three reference materials for Thromboplastins BCR No.147,148 and 149 1984
WHOのテクニカルレポート(1983、Geneva、上記①)はトロンボプラスチンにおいてWHOのIRPを使用する 場合の方法を勧告(Recommended)しています。その中で、まず、 WHOのIRPに付けられたISI値は主要 な10の研究施設で検定がなされていること、そして、経口抗凝固薬治療のコントロールにおいて、検査室で較 正(Calibration)する際にはWHOが推奨するIPRを使用すべきであること、が記されています。そして、 WHOのIRPには2種類のトロンボプラスチンがあり、トロンボプラスチン注出のみのもの(Human and Rabbit Brain)と結合型(Combined = Bovine注出物に F.Ⅰ+F.Ⅴ+Caを添加したもの)があり、較正の 対象となるトロンボプラスチン試薬(WRP)は較正の最適精度を求めるためにお互いに類似したトロンボプ ラスチンを使って実施すべきであるとしています。最初に最適なIRPを使用してWRPの較正をおこない、 次に、ロット毎にISI値を設定する作業を実施せよ――としています。
次に、較正の方法が記載されている部分について述べます。
較正(Calibration)においては、健常人2名とワーファリン投与患者6名の検体が使用されます。 (この数はクマリン誘発凝固欠損の個人差が大きいと言う点を考慮して算出されている。)健常人は男女各1名 であり、ワーファリン投与患者は少なくとも6週間に渡って経口抗凝固療法がおこなわれた安定した6名の検体 を使用すること、それぞれの検体はNormal 1~2、Patients 1~6と言う番号が付けられ、次の順序で、 較正作業は10日間おこなわれるべきであるとしています。
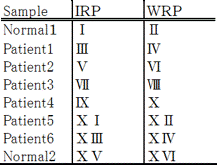
採血の方法は、血液9容に対し、0.109mol/Lのクエン酸3ナトリウム溶液を1容混合します。血液はプラス チック(ポリポロピレンなど)またはシリコンコートしたシリンジで採取し、同様の凝固活性化を生じせしめな い器具に移します。血液は直ちに遠心分離(2500rpm×5min、室温)し、上清を試験管に移し、栓をし、測定ま で室温に静置する。採血後、4時間以内に測定を終了させます。 IRPは、使用方法に従って準備し、溶解後は2時間以内に測定を終了させます。数日間実施する場合には IRPとWRPの順序を交互にして測定します。
測定は2重測定ではなく、シングルでおこないます。理由はシングル測定で異常が発生した場合には異常 なデータをグラフから読みとって排除できますが、2重測定した場合には残差誤差がわずかであり異常の発見が 遅れてしまうこと、また、測定に時間がかかり、検体の経時変化の影響を受ける可能性が増すため、早く連続し ておこなえるようにするためです。
測定に際し、エンドポイントはIRPの場合、手と目で見て確かめます。WRPの場合は用手法 or半自動、 または全自動の機械で測定して構わない。精密度3%以下、正確度(Variation) 5%以下であるべきです。
*国際標準トロンボプラスチンの国際比較の施設
(1) Hippocration Hospital, 1st Regional Transfusion Centre, Athens, Greece.
(2) Hospital de la Santa Creu I Sant Pau, Servei d’Hematologia, Barcelona, Spain.
(3) Centre Hospitalo-Universitaore de Paris, Laboratoire Central d’Hematologie de I’Hotel-Dieu, Paris, France.
(4) Instituto Nacional de Saude, Department of Haematology, Lisbon, Portugal.
(5) Ospedale Regionale di Parma, 5a Divisione Medicina Generale, Parma, Italy.
(6) Ribe Coubty Hospital, Department of Clinical Chemistry, Section Coagulation and Fibrinolysis, Esbjerg, Denmark.
(7) Thame Thrombosis and Haemostasis Research Foundation, Thame, Oxon, United Kingdom.
(8) Withington Hospital, Thrombosis Reference Centre, Manchester, United, Kingdom.
(9) Katholike Universiteit Leuven, Department of Medical Research, Leuven, Belgium.
(10) Klinikum der Albert-Ludwigs-Universtat, Zentrale Einrichtung Tranfusionmedizin, Freiburg iBr., Federal Republic of Germany.
(11) Academisch Ziekenhuis Leiden, Department of Haematology, Haemostasis and Thrombosis Research Center, Leiden, The Netherlands.
設問(22): 較正対象となるトロンボプラスチン試薬(WRP)は較正の最適精度を
求めるためにお互いに類似したトロンボプラスチンを使って実施すべきか?
Yes or No
設問(23): WRPは、正常が2×10回測定されるとすると、ワーファリン検体は
6×10回測定されることによりISI値が設定される、か?
Yes or No
A-4-2 ICSH と ICTH の レポート
ICSHとICTHは共同して、Thrombosis and Haemostasis(1985)、前述文献②に、経口抗凝固療法 のコントロールにおけるPTのレポート方法について「勧告(Recommendation)」を掲載しています。この中で はWHOが標準トロンボプラスチンとするBCT/253を基準として、あるいはこの基準試薬に基づいて設定さ れた他のWHO標準トロンボプラスチンを用いて、経口抗凝固療法に適用される市販PT試薬にはこれらと比 較したISI値を付け、INRで報告することを勧告しています。すなわち、
① 経口抗凝固療法において使用される市販PT試薬は、WHOが標準トロンボプラスチンとして採用した試薬と 比較した傾き(Slope)をバッチ毎に示し、ISIとして表示すること。
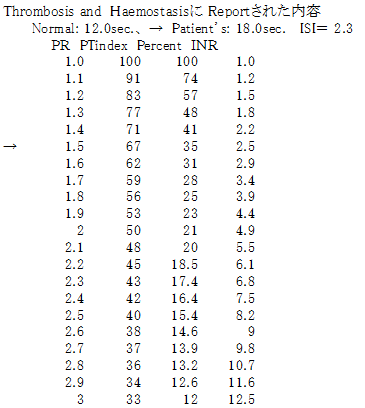
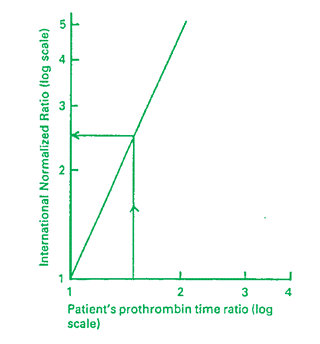
②市販PT試薬にはPT結果とINR値との関係を示した表あるいはグラフを添付すること。 例として下記の グラフと表が示されています。
A-4-3 ISI およびINR の 使用方法
まず、正常ヒト検体を準備する。正常ヒト検体はINR算出においてPRの底となり、結果を大きく左右 するので、非喫煙で健常な男女を選択し、可能な限り多数を集める。採血においては組織片の混入がないように 慎重に採取し、採血後は速やかに遠心分離して、測定に供します。
ワーファリン検体については、なるべく安定期の患者検体を上記と同方法で集め、測定します。PT試薬につ いては、メーカーの発行するISI値が付いているので、それを使用します。
①INR値を算出する場合
患者検体の測定秒数を正常検体の測定秒数で割り、PR値を算出する。
PR値にPT試薬のISI値を指数乗し、INR値を算出する。
患者検体が22.0秒で正常検体が11.0秒、ISI値が1.5であった場合には
INR=(22.0/11.0)1.5 = 2.828 となる。
②PT試薬のISI値を算出する場合
患者検体および正常検体を試薬Aで測定する。
同じ検体を試薬Bで測定する。
試薬Aで測定した時のデータを22.0秒、11.0秒とし、試薬AのISI値を1.5とする。
また、試薬Bで測定した時のデータを18.0秒、10.0秒とすると、
試薬BのISI値は、以下の計算により算出される。
(22.0/11.0)1.5 = INR = (18.0/10.0)ISIb
ISIb = 1.5 × Log(22.0/11.0) ÷ Log(18.0/10.0) = 1.768
③ 相関の傾きからPT試薬のISI値を算出する場合
なるべく多数の検体を試薬A、および、試薬Bで測定する。
個々の測定秒数を対数に変換する。(Log10を使用、Logeではない)
対数化されたデータを、Y軸に試薬A、X軸に試薬Bの相関をとり、一次回帰式を求める。
→ Y = aX + b
試薬AのISI値が1.5の場合、試薬BのISI値は、
ISIb = 1.5 × a
一次回帰式の傾きが1.2である場合には、
ISIb = 1.5 × 1.2 = 1.80
*比較する際の測定機器は同一のものを使用する。機器が異なる場合には機器としてのISI値を別途
算出して置くことが望ましい。
*また、現在入手できるPT試薬はメーカーが国際標準トロンボプラスチンと比較したISI値が表示され
ています。メーカーが示すISI値に疑問がある場合は、他メーカーの試薬と比較(できれば複数の)、然
る後に国際標準品を取り寄せて検討する方がより簡単であると思われます。
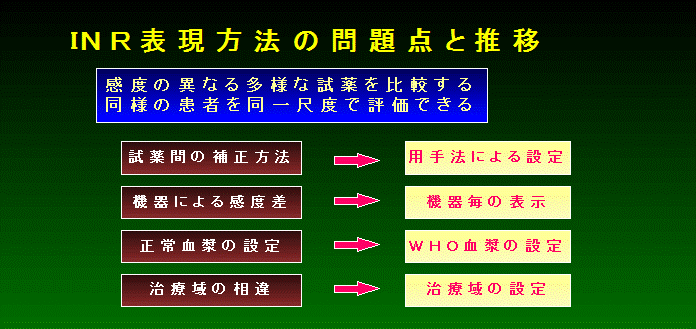
A-4-4 ISI/INRシステムの問題点
WHO、あるいはICSH・ICTHが勧告したISI/INRシステムは難産の末に提唱されたPTの 表現方法ですが、まだこのシステムには未完成の部分が残っており、これが故に実行上の問題となっています。 以下に問題点とその経過を示します。
第一には、ISI/INR法の実施に際し、測定方法に関する規定がない、あるいは不明確である点が挙げ
られます。WHOの勧告では国際標準品は用手法で測定し、その他の試薬は用手法または機器で測定するよう
説明していますが、それ以外の方法面での記載はありません。単に用手法と言っても同一とは考えずらい面が
あります。欧州での測定結果では同じ試薬を使っても15~16秒のデータとなるものが、本邦での測定結果では
11~12秒となると言う報告もあります。これに対し、国内では、取り敢えずは用手法で測定したものが基本で
あることを提議していますが、どの施設においても一定の結果となる訳ではありません。
第二には、機械毎に結果(感度)が異なる点を如何に補正すれば良いのか、と言う点です。上記第一の問題
点において“基本は用手法”と言うことに決定しましたが、機械毎の感度差が測定結果に与える影響は意外と
大きいこと(=ある機器をISI=1.0とすると、ISI=1.4と言う機器があること)が判り、機械毎の感度差は無
視できないものとなりました。そこで、試薬メーカーが表示するISI値表には少なくとも同社が表示し得る機器
毎のISI値を表示するように提案されました。この結果、各PT試薬にはメーカーで測定された値をもとに機器
毎のISI値が表示されるようになりました。が、ISI値の測定方法は決まった手技・手法が設定されていない現
状では、どうしてもメーカー毎に異なる傾向にあります。これを如何に統一的なものにするかと言う問題が残
ります。
第三には、検体に関する規定です。凝固検査では、一応、「正常ヒトプール血漿」を基準にしていますが、
「正常ヒトプール血漿」は集めた集団の特性により、凝固活性(秒数)も異なってくる傾向があります。正常
血漿の個々の平均値とプール血漿の測定値とでは、プール血漿の値が短縮傾向にあることも指摘されておりま
す。このため、各検査室で作製した「正常ヒトプール血漿」の出来方如何によって、PR値がブレたり、正確
度に疑問が生じたりすることになります。また、異常検体においても同様な状況であり、且つ、「安定期の
ワーファリン患者」の活性範囲については規定が示されていません。これを解決する手段として、近年、
“INR表示血漿”を使用することが提案されています。しかしながら、人種や食生活習慣の違う者(集団)
では、当然、正常域は異なってきますし、いわんや異常域においては感度的にも特性が異なってきます。
その結果、正常値のズレや異常域での値の違いはINR表示値に影響を与え、国際的に統一されたINR値で
比較検討できなくなります。
第四には、使用上の問題です。欧州ではワーファリンのモニタリングをPTだけで実施してきた経緯があり
ますが、本邦ではトロンボテストとPTを併用するか、ないしは使い分けてきた歴史があります。
このため、PTの検査によってワーファリンモニターをする習慣が少なく、INR検査依頼が少ない(あるい
は無い)と言った傾向のあることも事実です。また、治療面では軽度に凝固能を抑制する傾向(⇒INR値で
は2.0前後で治療する傾向)があり、ワーファリン治療に対する取り組み方の違いがあります。臨床医師サイド
での治療域を明確にし、且つ、検査リクエストを出してもらう必要があります。
第五には、凝固測定における測定手技の標準化が進んでいないと言う点です。上記一~四の如く、問題点が
解決されたとしても、基礎になるデータがバラバラであっては標準化の意味はありません。しかしながら、
凝固測定は施設毎に測定手技がまちまちであり、同一の試薬・機械・標準物質を使用していても施設間差は
極めて大きいのが現状です。正常or治療域においては正確なINR値を示し得る手法を規定することが必要で
あると考えられます。
その他、INR表示においてINR値が高いほどINR値が変動し易い性質がある点があります。またINR
値はISI値が大きい程、変動し易い変動し易い性質もあります。これらの点を踏まえ、多様な試薬に対応する
方法の規定も必要です。さらに、特にトロンボテストとPTとの関係を明確にすること、一律の計算方法では
適用できないと言う問題点に対し、今後、検討されるべき課題が多くあると思われます。
B ISI値設定方法、および管理方法
まず、当社標準PT試薬、および当社標準INR血漿の選択をおこないます。その方法としては、WHO
プロトコールに従い、正常ヒト血漿の測定、および、ワーファリン血漿の測定が大バッチで実施されます。
この際、使用される試薬はWHO標準試薬と次期候補の社内標準PT試薬です。次期候補の社内標準試薬は
事前の小バッチの検討で絞り込まれたものが使用されます。また、正常ヒト血漿は、社内健康診断時に健常人
(約200~300名)からクエン酸採血し、採血後2時間以内に個々の測定を実施します。同時に正常ヒト血漿は
プールされ、一部は凍結保存されて以降の各種の試験に使用されます。この作業により、正常ヒト血漿の平均
値と同等な秒数結果を示す凍結乾燥血漿(当社標準INR血漿)が選択されます。
次に、この作業と平行する形で約100検体のワーファリン検体が測定されます。測定結果は対数相関がとられ、
WHO標準品との一次回帰式の傾きから、次期候補の社内標準試薬のISI値が設定されます。
さらに、用手法・各種機器での測定結果から、対数相関をとり、測定に供した全試薬のISI値の変動が
計算されます。
以上の結果から、最も適正と考えられる試薬を選定し、これを当社標準PT試薬(STD)とします。この
試薬は製品系列毎に設定され、約1年間、運用されます。
次に設定された当社標準PT試薬、および当社標準INR血漿は精度管理部門、品質管理部門、およびその
他の部門に配布されます。精度管理はそれぞれブラインドされた2ヶ所以上の社内施設で、少なくとも1回/
週以上の頻度で実施されます。また、半年に1回以上の割合で、20名以上のプール血漿を作製して正常値の
検定をおこないます。品質管理は当社標準PT試薬、および当社標準INR血漿を使用して、各製品の検査
をおこないます。この結果、各製品に対しては統一された検査結果が表示され、且つ経時的変化を比較でき
るデータが得られることになります。
これらの日常的な使用においてデータ的異常が発生した場合には、直ちに原因究明と対策が実施されます。
一方、市場品質を監視する目的で、WHOのプロトコールに従ってISIの検定が実施されます。他社試薬
を始め、各種機器および機種間ISIの検定がおこなわれます。この結果は当社標準PT試薬・標準血漿の
選定方法の検討にフィードバックされると共に、機種間補正に使用されます。この作業を実施する部門は上記
の部門とは別であり、市場問題の発生に対して不定期に実施されます。
設問(24): ISI値は機器毎に異なる、か?
Yes or No
一定である、か?
Yes or No
C.凝固反応の理解
――技術解説から:プロトロンビン試薬の反応機序――
C-1 はじめに
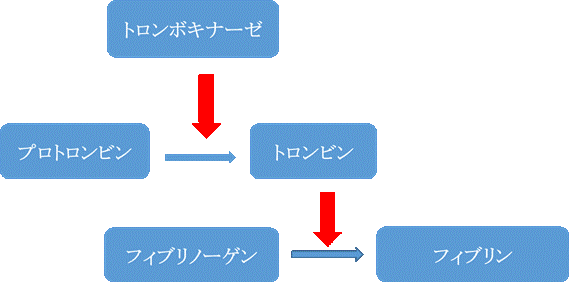
プロトロンビン時間測定(以下PT測定)は、1935年にA.J.Quick1)が1段法によるプロトロンビン 時間測定方法を考案したことに始まる。当時、一般的に理解されていた凝固反応系は極めて単純なもので (図1.)2)、トロンボキナーゼと称する物質が酵素前駆体であるプロトロンビン(凝固第Ⅱ因子)を 活性化酵素であるトロンビンに転化させる。さらにトロンビンはフィブリノーゲン(凝固第Ⅰ因子)を フィブリンに変えることによって、クロット(Clot)が形成されると考えられていた。ゆえに、プロト ロンビン時間測定とは簡易的なプロトロンビン測定法として提唱された。 A.J.Quickは提唱後、 1段法によるPT測定が凝固能を反映するか否かについて心配したが、幸いなことにPT測定は、今日 の画期的に変化した Water Flowの凝固反応系(図2.)で外因系を反映することが確認された。この ため、PT測定は血液凝固系の主要な検査項目として世界的に広く普及するようになった。
一方、 Quickが当時に使用した試薬は各種の組織をホモジナイズし、その抽出物の懸濁液であった。 当初は酵素と考えられていたためトロンボキナーゼと呼称されたが、後にこれが酵素ではなく、国際的な 名称も組織トロンボプラスチン(=第Ⅲ因子)と付けられた。しかし、さらにその後、各種の研究により 反応の本質が明らかにされ、組織因子(ティッシュファクター=TF)と命名された3)。今日では、TF は蛋白質構造が決定され4)、遺伝子工学的に製造されるようになっている。
これらの発展に伴い、PT試薬の構造、反応の機序が解明されてきている。このため、従来考えられ てきた概念とは趣を異にしている。PTの反応系、ひいては凝固反応系全体の見方を変えなくては理解でき ない状況が発生しつつある。本技術解説ではPT試薬の成分・構造、および反応機序についてこれまでの 情報をまとめてみたい。
C-2 PTの反応機序
C-2-1 PT試薬の製造方法
一般的に、動物由来(ヒト以外)PT試薬の製造方法は図3.に示す工程で製造されるが、基本的
にはA.J.Quickが開発した方法と大差はない。かつては国内の実験室においてもこの工程にしたがって、
ウサギなどの動物脳を調達し、PT試薬を調製することがなされていたが、現在は臨床検査の場で自家調製
することは希で、メーカーで作製された商品を使用するのが一般的である。
ただ、近年においてはメーカーの作業も分業化され、図3.の①~③の作業(=つまり、ウサギなどの
動物調達・脳などの摘出・アセトンパウダーの作製する工程)は専門メーカーによっておこなわれ、PT
試薬のメーカーは購入したアセトンパウダーを原料としてPT試薬を製造するのが一般的である。
① 各種動物(ウサギなど)の各種組織(脳など)を摘出する。
② アセトン中でホモジナイズし、脱脂・脱水する。
③ 乾燥させ、粉末とする(アセトンパウダーを作製する)。
④ 生理食塩水などの溶液に懸濁する。
⑤ 遠心分離し、上清画分(懸濁液)を得る。
⑥ その懸濁液をPT試薬とする。
あるいは凍結乾燥して保存する。使用時には水を加えて溶解する。
C-2-2 TFの構造
TPの抽出工程(図3.の④~⑤の作業)は、従来は組織片を得る作業と考えられていたため、
遠心分離の工程ではウサギ脳粉砕物の大きな破片を取り除き、試薬として有効な画分を得るために「ゆる
やかな遠心」が必要と考えられてきた。PT試薬の有効成分が組織片であると考えるとこれで良い。
が、TFであると考えると、この工程は以下のように複雑である。
つまり、これらの工程において、脳などの組織・細胞内にあったTFは破砕されてTFの一部に
細胞表面のリン脂質断片を付けた蛋白片となる。そして乾燥後、再度生理食塩水などに分散されると、
TFおよびリン脂質断片は液中に分散される。リン脂質断片の一部は凝集し、ミセル(=マイクロ
パーティクル:Mp)を形成する5)。このマイクロパーティクルは、TFが活性を発揮しうる形に
構成された第Ⅶ因子活性型マイクロパーティクル(以下Mp+)と、第Ⅶ因子活性化作用を持たない
マイクロパーティクル(以下Mp-)が形成される。
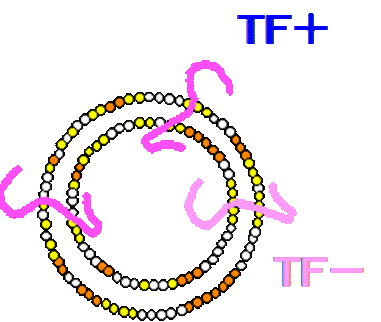
図4.マイクロパーティクルの構造模式図
このような工程を経ると、必然的にかなりの確率でMp+が形成される。そればかりか、TFと
分散性の良いリン脂質があれば、どのような動物種からも、あるいはどのような組織からでもMp+を
調製でき、PT試薬として利用できる。特に脳組織がPT試薬に適するのは、ホミジナイズによる分散
性が良く、且つ、リン脂質に富んでいることに依る。
ほとんどのMp+は、細胞膜と同様に、リン脂質二重層の形に再構成される。Mp+の大きさは分散
させた溶液中の撹拌力によって異なるが、一般的には0.1~1.0μmである。Mp+内部は空洞であるが、
再構成時の液体で充満される(図4.)。
この状態でTFはCaを添加してそのまま使用されるか、あるいは一旦凍結乾燥して使用される(図
3.-⑤)。凍結乾燥は長期保存を目的としておこなわれる。凍結乾燥では、反応環境を整え、凍結乾
燥のショックを和らげる目的で、緩衝剤や安定化剤を混合した溶液を試薬構成液として使用される。
Mp+が構成されたとする概念では、長期保存のためにはMp+内部の水分を除去する必要があること、
一旦再構成されたMp+内部の水分を抜くには強度の真空乾燥が必要であること、また試薬構成液は
塩析物がMp+の膜構造を破壊することのないように、また同時に溶解時点で活性を有するMp+に
復元されるように成分を調製することが必要である、と言った見解となる。このため、各種の製造技術
が必要である。
なお、最近では、遺伝子工学によって調製されたTF蛋白質とリン脂質からPT試薬が製造される。
この場合は正しく上記の概念(=Mp+形成)が実行されている。
C-2-3 酵素反応の段階
PTの反応は多段にわたる酵素反応と、フィブリンの形成反応の2つから成る。最初に起こるのは
酵素反応で、これは反応はMp+の分子表面上で進む。(図5.)
血液から供給された凝固第Ⅶ因子(F.Ⅶ)はCa2+を介してリン脂質膜と結合すると共に、一端は
Mpのリン脂質膜の一部から表面に突出したTFと結合する。これにより活性化凝固第Ⅶ因子(F.Ⅶa)
が生成されると考えられている。次にF.Ⅶaは凝固第Ⅹ因子(F. Ⅹ)に作用し、F.Ⅹaを生成する。
さらに、リン脂質膜上においてF.Ⅹa・F.Ⅴa複合体(以下Mp・F複合体)が形成され、F.Ⅱを分解し、
トロンビンを生成するようになる。この過程において、F.ⅦaはTFと結合した場所で、あるいは再度
浮遊した状態でF.ⅩをF.Ⅹaに変える。 F.Ⅶ、F.ⅩおよびF.Ⅱは Ca2+を介してリン脂質膜と
結合するが、どこにでも結合するのではなく、リン脂質膜上の負荷電性の高い部分(=PEまたはPS
の集中した部分と思われる)にまずCaが結合し6)、その上に各凝固因子が分子N末端側のγ-カルボ
キシグルタミン残基を介して結合する。一方、F.Ⅴは Ca2+を介して結合するのではなく、リン脂
質膜に埋もれる形で結合(あるいは膜が変形して結合)するため、荷電性のないリン脂質部分(=PCと
考えられる)に結合する。
複数の凝固因子とMp+が複合体を形成すれば、酵素と基質の分子量の差が大きくなり、分子会合の
確率が高まるので、一連の反応性はさらに向上することになる。また、Mp・F複合体は巨大分子とな
るために構造破壊の危険性が極端に低下し、高稼働率でしかも安定的な状態でトロンビンを生成する
「工場」となる。
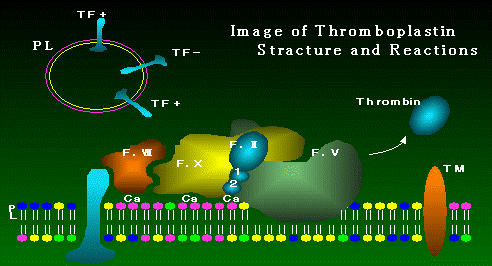
図5.外因系の反応模式図
凝固因子とMp+とによって、活性を発揮する有効なMp・F複合体「工場」が常にすべて形成さ
れる訳ではない。 Mp・F複合体の形成は、血液から供給された各種の凝固因子(FⅦ・FⅩ・FⅤ)
の量と、Mp+(およびMp-)の量によって影響され、マトリックスに組み合わされた不完全な幾種類
ものMp・F複合体が形成され、その中で有効に組み合わされたMp・F複合体のみが活性を発揮する関
係である。有効なMp・F複合体が形成される確率は正しくポアソン分布に従う。
これらの要素の中で最も重要なものはMp-の存在である。PT試薬は製造工程においてアセトンパウ
ダーを再浮遊させた懸濁液を使用するため、Mp-が必ず混入する。Mp-はMp+と同様に凝固因子を
吸着し、不活性型のMp・F複合体を形成する。このため、有効なMp・F複合体が形成される確率は
Mp-の多少により大きく左右される。(図7.参照)
C-2-4 酵素反応とCa
血液から供給される凝固因子はクエン酸ナトリウムによって Ca2+がキレートされており、
Ca2+欠乏の状態にある。PT試薬中に Ca2+が存在しないと凝固因子はMp+(またはMp-)
に結合できず、各凝固因子の活性化が非常に困難になる。このため、 Mp・F複合体を形成するには
Ca2+は不可欠である。
Ca2+とMp+、さらに各凝固因子が最適条件下で混合された状態では、有効なMp・F複合体が
多量に形成されるため、反応量が増し、したがって、凝固時間は短くなる。一方、 Ca2+が不足した
状態では、 Ca2+量に応じたMp・F複合体が形成されるため、凝固反応量(速度)は低下し、凝固
時間は延長する傾向を示す。
逆に、 Ca2+が過剰な状態では、 Ca2+はリン脂質表面に結合するだけでなく、溶液中に浮遊
する各凝固因子にも結合する。この状態では、本来複合体を形成すべきものがお互いに反発するために
Mp・F複合体を形成しにくくなる。そのために、凝固反応量(速度)は低下する。ただ、溶液中に
浮遊する Ca2+が結合した凝固因子であってもゆるやかには反応するので、 Ca2+不足の状態に
比べゆるやかな延長傾向を示す。
このようにPT測定の反応溶液中には最適な Ca2+が必要である(図6.)が、この必要量は抽出
液に残存する Ca2+の状態によって、さらにはMp+(またはMp-)の量により異なる。つまり、
製造の条件や、試薬の濃淡・試薬量の変動によっても、さらには試料中の抗凝固剤の添加濃度によって
も変わってくる。したがって、試薬メーカー、および製造ロットが異なれば Ca2+の添加量もそれに
合わせた量が必要となる。
反応溶液中には適切な量のCa量が存在する必要があります。しかし、見方を変えれば、このことは
Caの量を増加/減少させれば、反応量が異なることを示しています。つまり、多量にCaが存在する
場合には正常に凝固反応を実行できるマイクロパーティクルの相対数は減少することになりますから、
反応性は弱まり、結果として感度の高い試薬に表現されることになります。採血時のクエン酸濃度が
異なれば同一の試薬であっても、感度(ISI値)は違ってくることになります。
また、上述のごとく、マイクロパーティクルのりん脂質構造によってCaの最適量は変わるのです
から、試薬(メーカー)が異なればCaの添加量もそれに合わせた量が必要となり、一定のあるいは
固定的なCa量と言うものはありません。試薬(メーカー)毎にCa量は異なって当然です。製造技術
的にはCaがりん脂質に結合する力より弱く、凝固因子に結合する力より強い緩衝物質があれば、液中
にCaをストックしておくことが可能ですから、反応系には常に適正な量のCaを供給できるシステム
になります。その意味では緩衝剤はPHだけでなく、Ca供給の面で重要な作用となります。
C-2-5 クロット形成反応の段階
Mp・F複合体により生成したトロンビンは液中へ遊離され、フィブリノーゲンに作用してフィ
ブリンモノマーを生成する反応を開始する。
最初の段階では生成したトロンビンのほとんどはアンチトロンビン物質による阻害を受けて、失活
する。産生初期のトロンビンの70~80%はAT-Ⅲによって活性を失い、またフィブリノーゲン
やプロテインCなどの作用基質によっても失活される。また、20~30%のトロンビンは生成され
たフィブリンに吸着されることでも不活化される。その他、トロンビンは様々なアンチトロンビン物質
によっても失活すると考えられる。7)PT測定においてはこれらアンチトロンビン物質による消化が
済んだ後、フィブリンモノマーが加速的に産生される。
産生されたフィブリンモノマーは特定濃度(閾値)に達した時点より分子重合を開始し、フィブリン
塊を形成する。フィブリンモノマーの重合は非酵素的である。フィブリンモノマーの重合は、反応環境、
つまり、PH・電気伝導度・浸透圧・挟雑物質の存在量によってフィブリンポリマーの形成反応速度は
量的にも質的にも影響を受ける8)、9)、10)。一般的に中性で、塩濃度が低い状態での重合度が
速い。フィブリンポリマーの形成が発現されると、これに伴なって、透過光・散乱光の変化や粘度の
変化、あるいはフィブリン繊維が認められるようになる。
このようにPTの測定において、フィブリン形成までの各種の反応がn(ナノ)秒単位で進行している
にもかかわらず、我々が検知できるようになるまでに約7~8秒を要するのは、効果的に酵素作用を発
揮できるトロンビンの量が必要であることと、それに続く重合開始に必要な濃度のフィブリンモノマー
の量の確保に時間を費やすためである。
C-2-6 PT試薬の感度
PT試薬の感度は正常検体を測定した場合の凝固時間(正常値)と、凝固因子量の少ない異常検体
を測定した場合の凝固時間(異常値)によって決定される。正常値が示す凝固時間については特に規定
はないが、異常域での延長度が大きい場合には「感度の高い試薬」として位置付けられる。感度は総合
的な試薬特性を表現しており、諸種の要因により影響される。
たとえば、Mp+量、凝固因子量、Ca量などが最適な場合には凝固時間は正常値・異常値共に短く、
従って試薬感度としては鈍くなる。しかし、これらの量に過不足が生じた場合には試薬感度は急激に変
化し、高い感度を示すことになる。我々の実験(図7.)では通常のPT試薬にMp-を1%(or5%)
添加すると、ISI値1.70は1.467(or1.263)に下がり、また、PT試薬中のTF(Mp+、およびM
p-)濃度を1倍~10倍に変更した場合(図8.)にはISI値は1.00まで下がり、著しい感度の向
上が認められている。この意味では感度が高いと言うことは成分の適正なバランスのとれた試薬とは言
い難く、 ISI値が1.Oに近いと言うことは凝固因子・プロテアーゼ反応において阻害要因の大きい試
薬であることを窺わせる。
一方、凝固反応の場であるマイクロパーティクル(Mp)のリン脂質部分はPE・PS・PC・PA
などで構成される。これらの各成分は個々に荷電性が異なるため、荷電性の高い成分が多く混じるMpでは
Ca結合力が高く、したがってF.ⅦやF.Ⅹに対するの高い結合が期待できる。つまり、リン脂質成分
の差によって、PT試薬特性・感度に差が生じることになる。現在市販されているPT試薬におけるリン
脂質成分の差とは、原料の差や製法の差である。使用されている動物種としてはウサギ・ウシ・ウマ・
サル・ヒトなどであり、組織としては脳・肺・胎盤などが挙げられるが、これらに由来するリン脂質成分
に差があり、したがって上述の如く、感度にも差異が生じることになる。
ごく最近に至ってはTF蛋白質を遺伝子工学的に大腸菌などに合成させ、リン脂質部分は人工的に調合
された構成成分とし、この両者を再構成させることでよい均一性の高い(?)PT試薬が製造されるよう
になった。これは、リコンビナント試薬として販売されている。ただ、このように合成されたMpではMp
作製後にTFを付着させなければならないために”浮遊”するTFが存在して凝固時間が短くなる点、
また生体内の反応ではMpはリン脂質のみでなくコラーゲンなどの物質も存在しており、これらの物質が
凝固反応を制御している面もある。ゆえに人工的に合成されたMpでは生体内の凝固能を完全に反映した
ものとは言えない面もある。
C-3 おわりに
以上、PT測定における凝固反応の概念について述べた。凝固反応は反応の場であるMp+と凝固
因子との椅子取りゲームに例えることができる。Caを介して椅子にたどりついた凝固因子はMp・F
複合体を形成し、凝固反応が促進される。その間にはCaが重要な役目を果たすことを説明した。しかし、
PT測定は以下の特徴的な条件で実施されていることも留意して置く必要がある。
① PT測定では高濃度の蛋白質溶液中で行われる点である。一般的に実施される生化学検査や免疫分
野の測定では反応液中の被検試料濃度はかなり低い。が、PT測定ではこれが1:2であり、せいぜい
3倍にしか希釈されない。そのため、Mp+と凝固因子蛋白質との会合確率の点で立体阻害を生じる反応
環境となっている。このため、会合条件を安定的に保つには強度な撹拌が必要であり、撹拌が弱い場合には
データへの影響に注意しなければならない。
② PT測定では多段に渡るプロテアーゼ反応の結果として凝固能が測定されている。一般的に凝固因子
は極めて不安定であるため、被検体の取扱い方法によって重大な測定誤差を発生する。しかも被検試料中
には他種類の抑制物質が共存するため、異常データの成績判断には注意を要する。
③ フィブリンクロットの形成は自然重合でおこなわれるため、反応環境が大きく影響する点である。
反応溶液中の PH・ 電気伝導度・ 浸透圧・ およびその他の測定条件が影響する。しかも反応が進むに
つれてgelはsolに変化し、重合反応の環境も変化する。凝固終末点の検出方法は種々の原理・方法が
考案されている13)が、仮に同一被検試料であったとしても機器の検出原理や解析方法によってまた様々
な結果が示される。
以上のようにPT測定は成分も反応様式も、また測定方法も複雑であり、その理解は難しい。しかし、 一方では標準化の検討も盛んにおこなわれている。14)、15)INR表示は国際的な規模で評価されて おり、もっとも期待できる方法であると考えられる。標準化が進展すれば凝固検査分野でのさらなる発展が 期待できる。
以上
4-C-4 文献
1) Quick,A.J., B.M.Stanly and F.W.Baneroft:A study of the coagulation defect in hemophilia and jundice. Am.J.Med.Sci.,190,501~511,1935
2) Morawitz,P:Beitrage zur Kennits der Blutgerinnung. Beit. Chem. Physiol.,5,133,1904
3) 福武勝博、浮田実:日本血液学全書11,出血性素因・基礎 P217、新版日本血液学全書刊行委員会編、丸善、1979
4) 濱本高義、中垣智弘、岩永貞昭:外因系血液凝固機構の進歩、血液・腫瘍科、Vol.34、No.6、502-511、1997
5) Papahadjopoulos,D.,C.Hougie and D.J.Hanahan:Influence of surface change of Phospholipids on their clot promoting activity. Proc.Soc.Exp.Biol.Med.,3,413,1962
6) 香川靖雄:生体膜と疾患の分子生物学 P480,南山堂、1993
7) 鈴木宏治:止血・血栓・線溶(松田道生、鈴木宏治編集)、P187-196,中外医学社、1994
8) 松田道生:フィブリノゲンの誘導体、とくに可溶性フィブリン(soluble fibrin)とDダイマー(D dimer)について、血栓止血,Vol.8,No.1,24-32,1997
9) 丹羽和紀:フィブリン繊維の形成機序に関する研究, 血栓止血,Vol.8,No.1,33-43,1997
10) 山角健介、田辺元、愛甲孝:合成ペプチドを応用した重合機転の解析、血栓止血Vol.8,No.2,75-83,1997
11) R.Bader、et.al.:Multicentric Evaluation of a New PT Reagent Based on Recombinant Human Tissue Factor and Synthetic Phospholipids、Thrombosis and Haemostasis、Vol.71、No.3.,292-299,1994
12) 高宮脩、百瀬正信、横瀬和哉:遺伝子組み替え型組織因子と臓器抽出物の組織因子を用いた第Ⅶ因子活性の比較、医学と薬学Vol.33、No.1,229-237,1995
13) 中野一司、東和也、西村辰志、北島勲、丸山征郎:凝固・線溶検査の自動化、日本臨床検査自動化学会誌、Vol.22、No.1.、3-8、1997
14)WHO Expert Committee on Biological Standardization、 33 report(WHO technical Report Series No. 687、1983
15) 鈴木節子ら:PT・TT標準化に関する研究、臨床病理Vol.45,No.4,321-327,1997
16) Clinical Laboratory Standards Institute. Collection, transport, and processing of blood specimens for testing plasma-based coagulation assays. Approved Guideline - Fourth Edition. CLSI document H21-A4. CLSI, 940 West Valley Road, Suite 1400. Wayne, Pennsylvania 19087-1898 USA, 2003.
17) **van den Besselaar AM, Hoekstra MM, Witteveen E, et al. Influence of Blood Collection Systems on the Prothrombin Time and International Sensitivity Index Determined With Human and Rabbit Thromboplastin Reagents. Am J Clin Pathol. 2007; 127: 724-9.
18) 金井正光編著:臨床検査法提要、第32版、p413(2005)、金原出版
19) 日本検査血液学会編:スタンダード検査血液学、p75-78、医歯薬出版株式会社(2003)
20)渥美達也:抗リン脂質抗体症候群、血栓止血誌12(6)p500~508(2001)
21)福武勝幸:凝固検査の標準化の現状:プロトロンビン時間(PT)、生物試料分析Vol.32、No.5(2009)